Presented by: Chrysa Koutsiouki MD

Edited by: Penelope Burle de Politis, MD

A 70-year-old man presented with a history of retinal vasculopathy and decreased vision in the left eye.
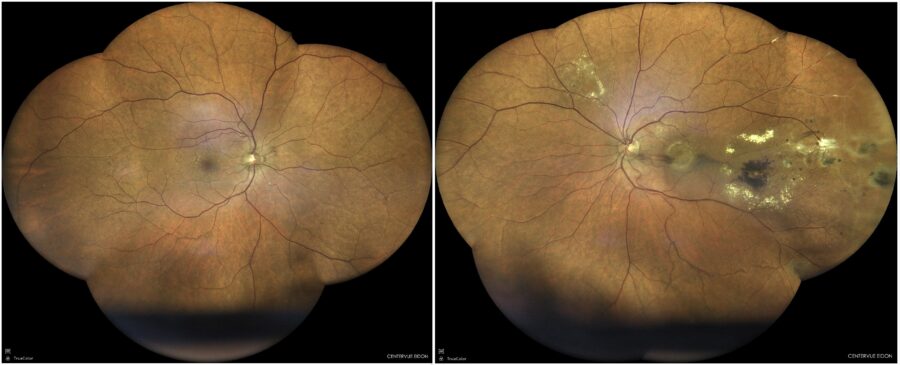
Figure 1: Fundus color photograph (iCare confocal high-resolution ultra-widefield Truecolor Centervue EIDON®) showing macular edema, periphlebitis and circinate hard exudates in the left eye.
Case History
A 70-year-old Caucasian man was referred for investigation of slowly progressive visual loss in the left eye (LE) over the course of 10 years. His past medical history was unremarkable. His family medical history was negative for ophthalmic diseases. Upon examination, his corrected distance visual acuity (CDVA) was 8/10 in the right eye (RE) and 4/10 in the LE, with a refractometry of +0.50D and +0.75-0.50@165°, respectively. There were no signs of uveitis in either eye. Intraocular pressure (IOP) was within the normal range bilaterally. Fundus examination of the RE was normal, while the LE presented macular edema, periphlebitis and circinate hard exudates (Figure 1).
Spectral-domain optical coherence tomography (SD-OCT) and OCT-A (optical coherence tomography angiography) revealed drusenoid changes in the RE and diffuse intraretinal fluid in the LE (Figures 2 and 3).
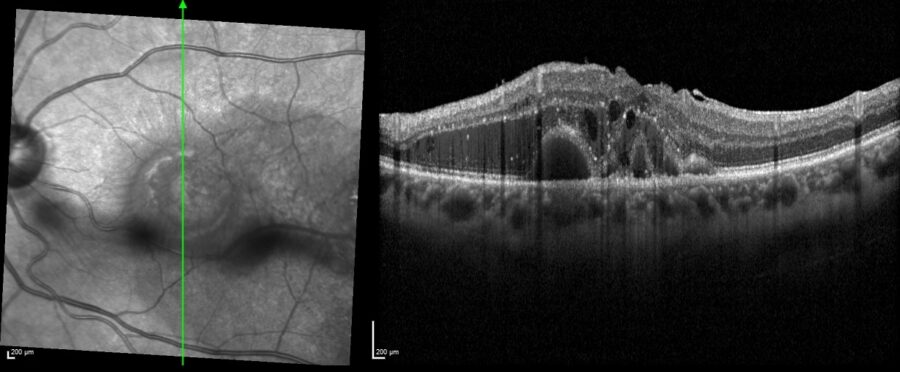
Figure 2: SD-OCT of the left eye (Spectralis, Heidelberg Engineering®) showing macular edema and intraretinal fluid.

Figure 3: OCT-A (Spectralis, Heidelberg Engineering®) displaying the angiographic aspect of the left macula at presentation.
Fundus fluorescein angiography (FFA) was normal in the RE and showed telangiectatic, aneurismatic, and ischemic changes in the LE, with no evidence of active vasculitis or a hot disc (Figure 4).
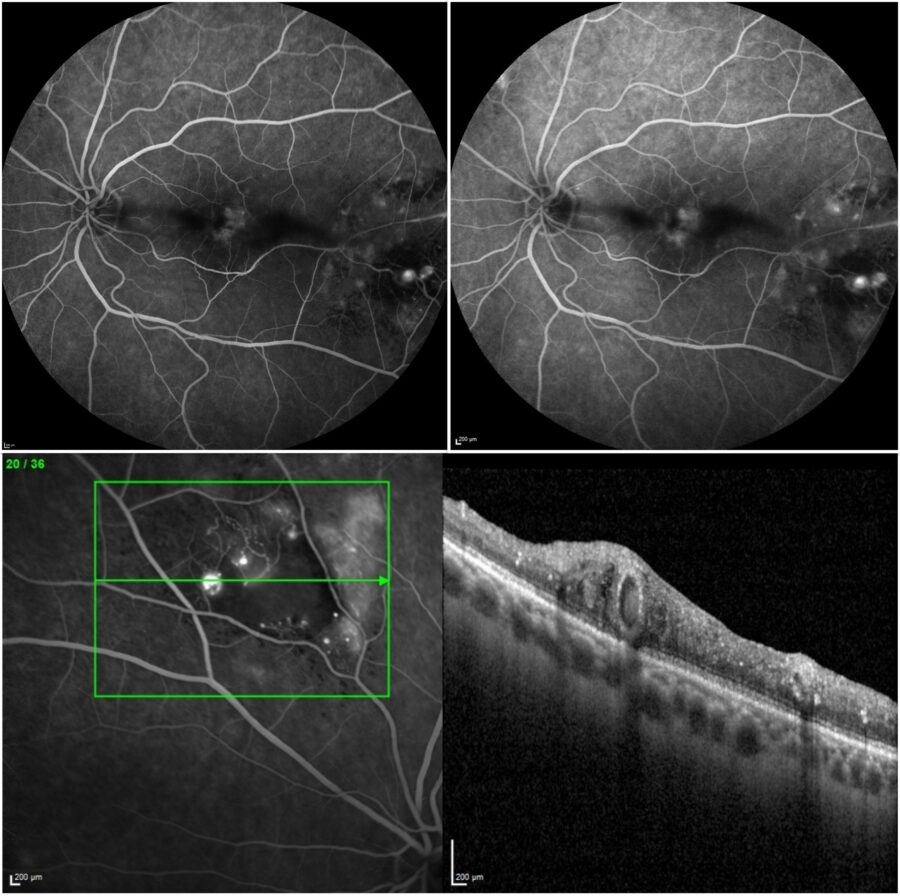
Figure 4: FFA (Spectralis, Heidelberg Engineering®) of the left eye showing telangiectatic, aneurismatic, and ischemic changes (upper left: early frame; upper right: late frame; bottom: sectoral magnification over the vascular lesions).
Additional History
The patient had been diagnosed with IRVAN syndrome (idiopathic retinal vasculitis, aneurysms, and neuroretinitis) in the LE ten years before. His recorded LE CDVA back then was 6/10. However, the vascular changes detected through the multimodal retinal imaging methods applied in the latest investigation favored the diagnosis of Coats’ disease rather than IRVAN.
The chosen treatment strategy consisted of intraocular dexamethasone implant and sectoral argon-laser retinal photocoagulation, with resolution of the macular edema and the intraretinal fluid over a 4-month follow-up period (Figure 5).
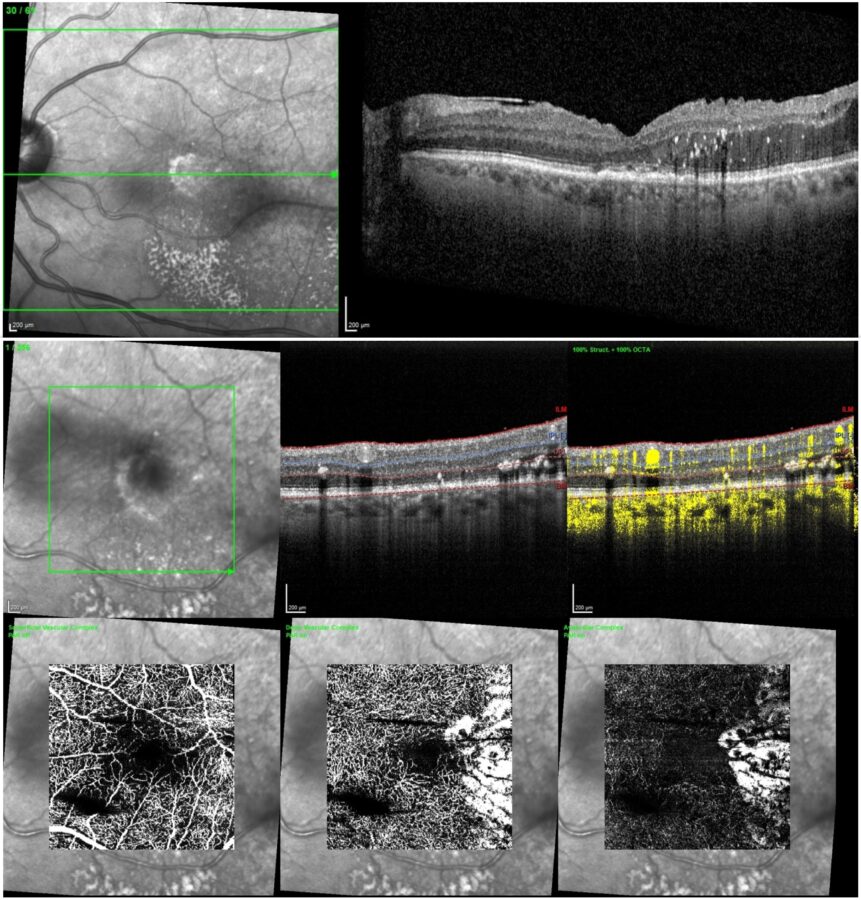
Figure 5: Post-treatment SD-OCT (top) and OCT-A (bottom) of the left eye showing resolution of the macular edema and intraretinal fluid (Spectralis, Heidelberg Engineering®).
Differential Diagnosis of Coats’ Disease in Adults
- IRVAN (idiopathic retinal vasculitis, aneurysms and neuroretinitis) syndrome
- Sarcoidosis
- Behcet’s disease
- Vasoproliferative tumors (e.g. retinal capillary hemangioma)
- Eales’ disease
- Sickle-cell retinopathy
- Leukemias
- Systemic lupus erythematosus
- Toxocariasis
Coats’ disease is characterized by retinal exudation and telangiectatic changes. When diagnosed late in life, Coats’ disease must be considered after the exclusion of other possible conditions. Unilaterality and the presence of abnormal capillary bed in the absence of vasculitis are suggestive of Coats’. A Coats’-like reaction may also occur in patients with ocular inflammation, infection, retinal degeneration, and following radiation therapy.
Discussion and Literature
Coats’ disease is an idiopathic, sporadic (non-hereditary) condition, first described by Coats in 1908 as a retinal telangiectatic and aneurysmal disease associated with retinal exudation. Also referred to as idiopathic peripheral retinal telangiectasia (IPT), it typically presents in young male boys, usually younger than five years of age. More than 75% of patients are male, and 95% of cases are unilateral. There is no clear racial predilection.
The complete pathogenesis of Coats’ disease remains unclear but involves breaking of the blood-retinal barrier, increasing vascular permeability, and presence of abnormal pericytes leading to weakness of the vascular wall. There have been reports of localized mutations in proteins controlling retinal angiogenesis.
The clinical presentation in Coats’ disease varies. Some patients may remain asymptomatic and be diagnosed by chance. Decreased vision is the most common presenting complaint. Some patients notice a defect in the visual field. Anterior segment signs such as those of uveitis may be present in advanced cases. While observation is a reasonable initial management strategy, modern multimodal imaging allows the detection of even subtle vascular changes.
Coats’ disease can be classified and staged using the Shields method (following the 2000 Proctor Lecture): Stage 1, retinal telangiectasia only; Stage 2, combined with retinal exudation (stage 2A, extrafoveal exudation; 3A2 foveal exudation); Stage 3, exudative retinal detachment (stage 3A, subtotal detachment; 3A1, extrafoveal only; 3A2 foveal detachment); stage 3B, total retinal detachment; Stage 4: total retinal detachment and glaucoma; Stage 5, advanced end-stage disease.
Treatment protocols depend on the age at diagnosis. For Coats’ disease diagnosed late in life, the treatment regimen must be case-tailored, considering disease presentation, staging and follow-up response. Combinations of intravitreal anti-VEGF, retinal photocoagulation and cryotherapy are the current mainstays. Intravitreal corticosteroids have been used to address macular edema and subretinal fluid. Epiretinal membrane and macular hole are possible long-term complications and must be managed accordingly.
Keep in mind
- Coats’ disease is mainly a vascular retinopathy of young boys. Cases presenting in adulthood usually feature less extensive involvement and more favorable outcomes.
- Because most cases of Coats’ disease are unilateral, other conditions must be considered for bilateral disease.
- When diagnosed late in life, Coats’ disease is managed on a case-based approach, according to disease severity and response to therapy.
References
- Mandura RA & Alqahtani AS (2021). Coats’ Disease Diagnosed During Adulthood. Cureus, 13(7), e16303. https://doi.org/10.7759/cureus.16303
- Ali Khan H, Ali Khan Q, Shahzad MA, Awan MA, Khan N, Jahangir S, Shaheen F, Wali K, Rodman J, Pizzimenti J & Saatci AO (2022). Comprehensive overview of IRVAN syndrome: a structured review of Case Reports and Case Series. Therapeutic advances in ophthalmology, 14, 25158414211070880. https://doi.org/10.1177/25158414211070880
- Ramli Norlina & Mimiwati Zahari. (2007). IRVAN syndrome. Philippine journal of ophthalmology. 14. 64-66. https://paojournal.com/article/irvan-syndrome/
- Gawęcki M (2021). Idiopathic Peripheral Retinal Telangiectasia in Adults: A Case Series and Literature Review. Journal of clinical medicine, 10(8), 1767. https://doi.org/10.3390/jcm10081767
- Ashkenazy N, Acon D, Kalavar M & Berrocal AM (2021). Optical coherence tomography angiography and multimodal imaging in the management of Coats’ disease. American journal of ophthalmology case reports, 23, 101177. https://doi.org/10.1016/j.ajoc.2021.101177
- Sen M, Shields CL, Honavar SG & Shields JA (2019). Coats disease: An overview of classification, management and outcomes. Indian journal of ophthalmology, 67(6), 763–771. https://doi.org/10.4103/ijo.IJO_841_19
- Kumar K, Raj P, Chandnani N & Agarwal A (2019). Intravitreal dexamethasone implant with retinal photocoagulation for adult-onset Coats’ disease. International ophthalmology, 39(2), 465–470. https://doi.org/10.1007/s10792-018-0827-0
- Li S, Deng G, Liu J, Ma Y & Lu H (2017). The effects of a treatment combination of anti-VEGF injections, laser coagulation and cryotherapy on patients with type 3 Coat’s disease. BMC ophthalmology, 17(1), 76. https://doi.org/10.1186/s12886-017-0469-4
- Grosso A, Pellegrini M, Cereda MG, Panico C, Staurenghi G & Sigler EJ (2015). Pearls and pitfalls in diagnosis and management of Coats’ disease. Retina (Philadelphia, Pa.), 35(4), 614–623. https://doi.org/10.1097/IAE.0000000000000485
- Otani T, Yasuda K, Aizawa N, Sakai F, Nakazawa T & Shimura M (2011). Over 10 years follow-up of Coats’ disease in adulthood. Clinical ophthalmology (Auckland, N.Z.), 5, 1729–1732. https://doi.org/10.2147/OPTH.S27938