Presented by: Stavrenia Koukoula MD, PhD & Theoni Panagiotoglou MD, PhD

Edited by: Penelope Burle de Politis, MD

A 16-year-old boy presented with a history of progressive bilateral visual loss since the age of 6.
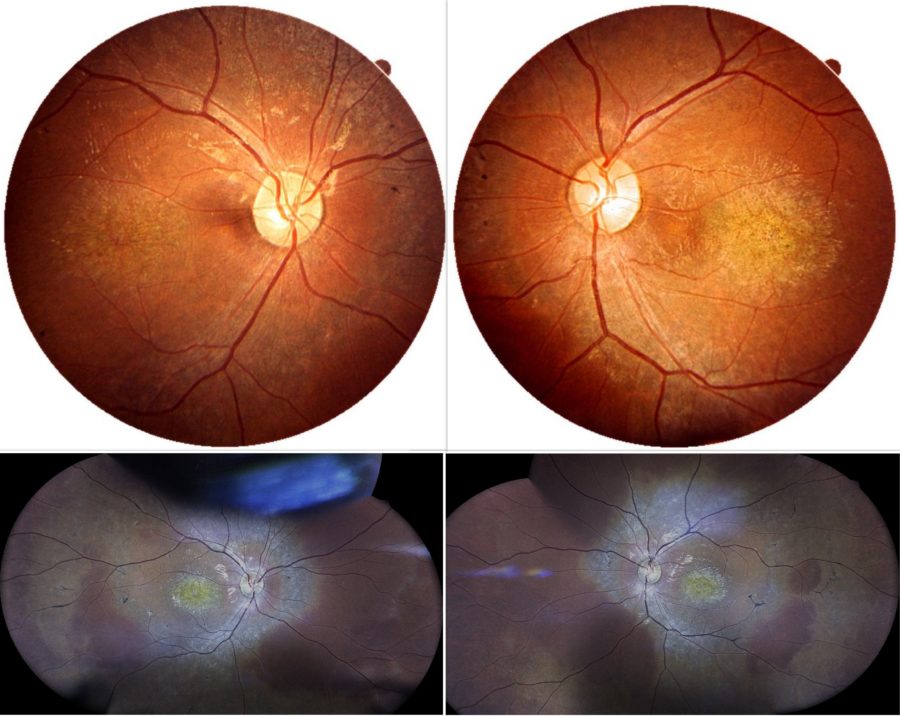
Figure 1: Color fundus photographs (top: REVO FC 80 Optopol Technology® retinal scan, courtesy of T. Panagiotoglou; bottom: iCare EIDON® widefield TrueColor confocal retinal mosaic scan) displaying retinal pigment deposits predominantly located at the macular region of both eyes.
Case History
A 16-year-old boy of Albanian descent presented with a history of progressive bilateral visual loss since the age of 6. The patient referred to intermittent right exophoria and complained of photophobia. Due to his vision deficit, he had recently dropped school. He was born full-term and had no systemic diseases except for chronic otitis. His family history was negative for ocular disorders or blindness, but his parents were distant consanguineous relatives. Under ophthalmological examination, his corrected distance visual acuity (CDVA) was 1/10 in the right eye (RE) and 2/10 in the left eye (LE). Total bilateral color blindness could be verified. Refractometry was -0.50@75° in the RE and -0.50@55° in the LE, and intraocular pressure was 14mmHg bilaterally. Anterior segment biomicroscopy was normal in both eyes (BE). Neither tropias nor relative afferent pupillary defect (RAPD) were present. Fundoscopy revealed pale discoloration of both macular regions, relatively pale optic discs, mild attenuation of the retinal vessels, and mid-peripheral bone spicule-like intraretinal pigmentation in BE (Figure 1).
Spectral domain optical coherence tomography (SD-OCT) evidenced a general macular atrophy, with foveal thinning with loss of photoreceptors, of ellipsoid zone (EZ line), of outer nuclear layer, and of RPE in BE (Figure 2).
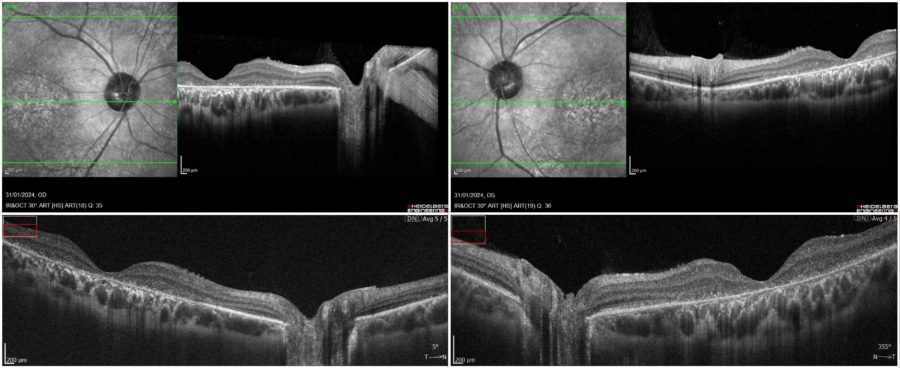
Figure 2: Posterior pole scans demonstrating foveal thinning and attenuation of the inner segment ellipsoid zone in both foveas. Top images, respectively RE and LE: infrared reflectance (IR) and spectral domain optical coherence tomography (SD-OCT, Heidelberg Engineering®). Bottom images, respectively RE and LE: SD-OCT (REVO FC 80 Optopol Technology®), courtesy of T. Panagiotoglou.
Blue-light fundus autofluorescence (BAF) imaging displayed various degrees of retinal atrophy (alternating areas of hypoautofluorescence and hyperautofluorescence; the more the atrophy of the retinal pigment epithelium (RPE), the more evident the hypoautofluorescence (Figure 3).
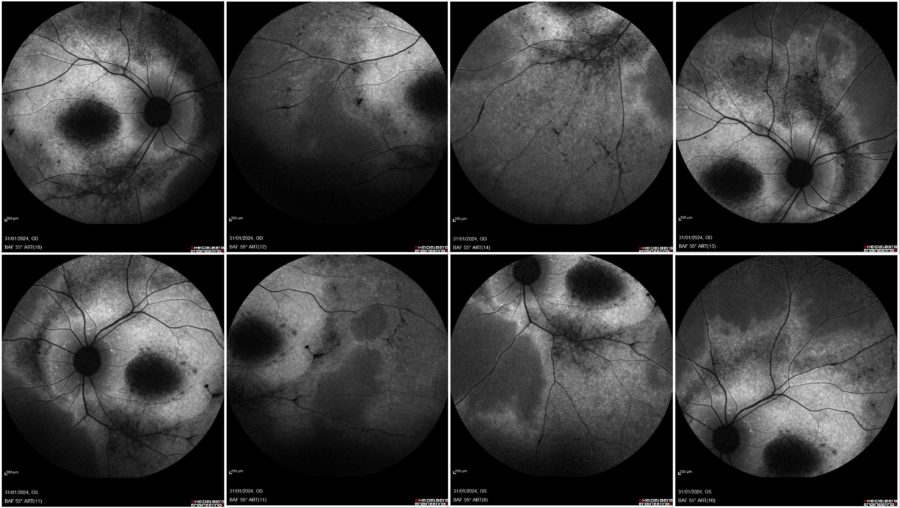
Figure 3: Blue-light fundus autofluorescence (BAF) photographs (Heidelberg Engineering®) illustrating a highly hypofluorescent macula and areas of RPE changes with speckled mid-peripheral hypoautofluorescence bilaterally (upper row: OD; lower row: OS; respectively at front, side, up and down gaze).
Additional History
Given the ocular history and fundus findings, the diagnostic hypothesis of cone-rod dystrophy was considered, hence the patient was referred for electrophysiological study of the retina. The electroretinogram (ERG) was consistent with generalized retinal dysfunction at the level of photoreceptors, both cones and rods, with macular involvement in BE (Figure 4).
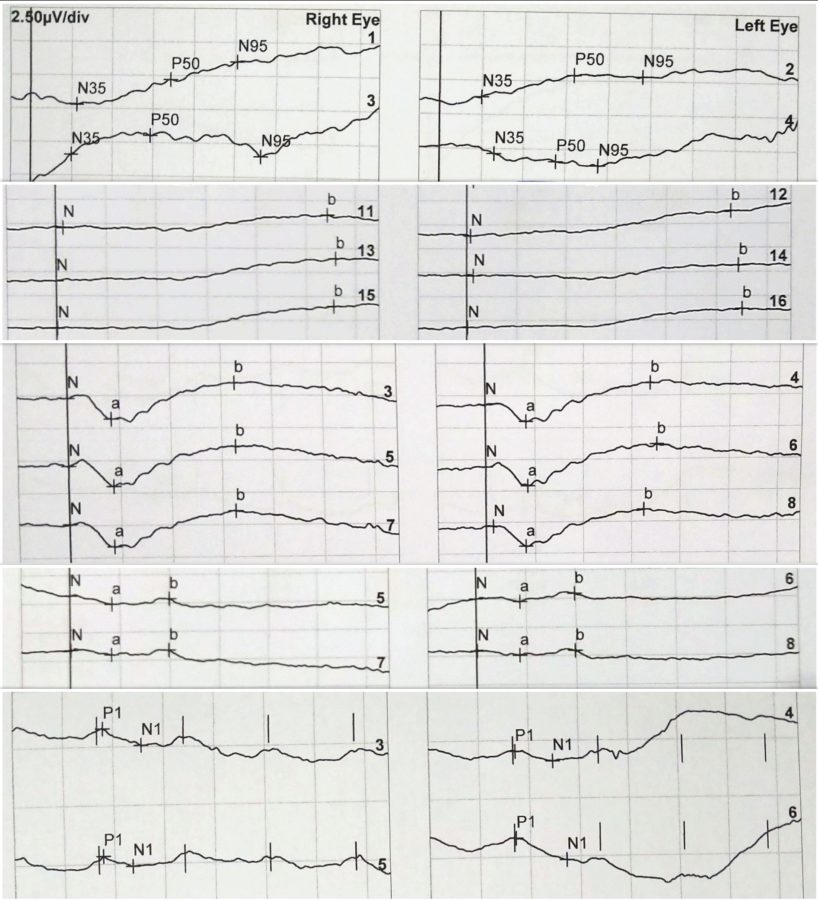
Figure 4: ERG records, respectively of RE and LE. From top to bottom: Pattern ERG (PERG): P50 undetectable to both standard and large field stimulus in BE. Full-field ERG (ffERG) protocols: rod-specific dark-adapted (DA 0,01) with b-wave amplitudes of 90μV on the RE and 80μV on the LE; bright flash dark-adapted (DA 10) with a- and b- wave peak times bilaterally subnormal and amplitudes of 50% the normal values; photopic light-adapted single flash (LA 3.0) with abnormal peak time bilaterally and a- and b- wave amplitudes respectively of 7μV and 15μV on the RE and 6μV and 13μV on the LE; 30Hz flicker (LA 3-30) peak times of 35-37ms (severely delayed), with amplitudes of 20-30μV (30% of normal) in BE.
Based on the ophthalmological examination, the multimodal image findings and the electrophysiological profile, the diagnosis of cone-rod dystrophy was confirmed.
Genetic counseling was given to the family and a multidisciplinary educational approach was advised.
Differential Diagnosis of Cone-Rod Dystrophy
- retinitis pigmentosa
- Stargardt’s disease
- Leber congenital amaurosis
- pure cone dystrophy
- isolated achromatopsia
- rubella retinopathy
- drug-induced retinopathy (e.g. antimalarials)
The differential diagnosis between cone-rod dystrophies and other retinal disorders is based on disease course, fundoscopic aspects and electrophysiological features. Accurate differentiation from overlapping photoreceptor dysfunctions is crucial for proper management.
Discussion and Literature
Cone-rod dystrophies (CRDs or CORDs) are inherited dystrophies belonging to the group of pigmentary retinopathies. The reported prevalence of CRDs is 1:40,000 (ten times less frequent than retinitis pigmentosa (RP or rod-cone dystrophy). In contrast to rod-cone dystrophies (RCDs), CRDs typically present primary cone involvement or, sometimes, concomitant loss of cones and rods. As a result, CRDs present first as macular disease, or diffuse retinopathy with predominant macular involvement. The classical symptoms of CRDs are decreased visual acuity, color vision deficits, photoaversion and decreased central visual field sensitivity, later followed by progressive peripheral vision loss and night blindness. The clinical course of CRDs is generally more severe and rapid than that of RCDs, leading to earlier legal blindness and disability.
CRDs are most frequently non-syndromic but may be part of syndromes such as Bardet Biedl, Spinocerebellar Ataxia Type 7 (SCA7), ectodermal diseases, and dysmorphic syndromes. Non-syndromic CRDs are genetically heterogeneous and all three patterns of mendelian inheritance are seen. The autosomal recessive (AR) pattern is the commonest (60–70%), followed by autosomal dominant (AD, 20–30%) and X-linked recessive (XL, 5%). Mutations in about 30 genes are involved in the pathogenesis of CRDs (about 20 are responsible for AR CRD and 10 for AD CRD). For AR CRD, the major implicated genes are ABCA4 (which causes Stargardt’s disease and 30 to 60% of AR CRD), CNGB3, KCNV2, and PDE6C. Dominant CRD is caused by a mutation in the GUCY2D and CRX genes. For X-linked cases, the affected gene is RPGR (which also causes about 2/3 of X-linked RP). There is some association between X-linked CRD and high myopia.
Two stages can be identified in the course of CRDs. In the first, the main symptom is decreased visual acuity, usually discovered at school, during the first decade of life. Patients often have a deviated gaze to project images on parafoveal less damaged regions. There is intense photophobia and a variable degree of dyschromatopsia. Night blindness is less prominent. Visual field testing shows central scotomas with peripheral sparing, hence patients have no difficulties moving. Fundus examination shows pigment deposits and various degrees of macular atrophy. Retinal vessels are normal or moderately attenuated. The optic disc is pale, particularly on the temporal side (macular fiber bundle). In the second stage, night blindness becomes more evident and peripheral visual loss progresses, compromising autonomous moving. Visual acuity continues to decrease to a level where reading is no longer possible. Nystagmus is often present. At this stage, patients are legally blind (visual acuity <1/20).
The diagnosis of CRDs is based on clinical history, fundus findings and electroretinogram. On the OCT of eyes with CRD, the IS/OS (ellipsoid) line is absent at the fovea. Fluorescein angiography and fundus autofluorescence show that the peripheral retina is also involved with heterogeneous fluorescence, but to a lesser extent than the macula. Since photoreceptors are distributed throughout the entire retina, imaging should not be limited to the posterior pole. Quantitative fundus AF (qAF) analysis (an indirect measure of lipofuscin within the RPE cell layer) may facilitate differential phenotypic diagnostics of CRDs. Characteristic electroretinogram (ERG) features are: implicit time (between a- and b-wave peaks) shift at the 30-Hz flicker cone responses; a- and b-wave single flash photopic response delayed (early), or with reduced amplitude (later); predominant involvement of photopic (cones) over scotopic (rods) responses. Molecular diagnosis can be made for certain genes but is not largely applied. Targeted next-generation sequencing (NGS) has been used to study the genotype-phenotype correlations in CRDs.
Visual prognosis is poor as CRDs lead to legal blindness in the majority of patients. Presently, there is no therapy to halt disease progression or restore vision. Management aims at slowing down the degenerative process (light protection, vitamin therapy), treating complications (cataract, macular edema, inflammation), and helping patients cope with the social and psychological impact of blindness. Filtrating spectacles can be used to minimize photophobia and low vision aids to improve functioning. Education must be adapted and carried out by a multiprofessional team. Genetic counseling is always advised. Ongoing research focuses on mechanisms of cone and rod cell death in these diseases so that their pathways can be better understood and potentially targeted for therapeutic intervention. Gene-based approaches are currently under study.
Keep in mind
- Cone-rod dystrophies are genetically heterogeneous pigmentary retinopathies that generally lead to legal blindness before adulthood.
- Low central visual acuity, photoaversion and poor color vision are the hallmarks of cone-rod dystrophy, nyctalopia being a later phase symptom.
- A precise diagnosis of cone-rod dystrophy requires electrophysiological study of the retina for adequate counseling and early rehabilitation.
References
- Hamel CP (2007). Cone rod dystrophies. Orphanet journal of rare diseases, 2, 7. https://doi.org/10.1186/1750-1172-2-7
- Thiadens AA, Phan TM, Zekveld-Vroon RC, Leroy BP, van den Born LI, Hoyng CB, Klaver CC, Writing Committee for the Cone Disorders Study Group Consortium, Roosing S, Pott JW, van Schooneveld MJ et al. (2012). Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology, 119(4), 819–826. https://www.sciencedirect.com/science/article/abs/pii/S0161642011009584
- Tsang SH & Sharma T (2018). Progressive Cone Dystrophy and Cone-Rod Dystrophy (XL, AD, and AR). Advances in experimental medicine and biology, 1085, 53–60. https://doi.org/10.1007/978-3-319-95046-4_12
- Georgiou M, Robson AG, Fujinami K, de Guimarães TAC, Fujinami-Yokokawa Y, Daich Varela M, Pontikos N, Kalitzeos A, Mahroo OA, Webster AR & Michaelides M (2024). Phenotyping and genotyping inherited retinal diseases: Molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, leber congenital amaurosis, and cone dysfunction syndromes. Progress in retinal and eye research, 101244. Advance online publication. https://doi.org/10.1016/j.preteyeres.2024.101244
- Oishi A, Oishi M, Ogino K, Morooka S & Yoshimura N (2016). Wide-Field Fundus Autofluorescence for Retinitis Pigmentosa and Cone/Cone-Rod Dystrophy. Advances in experimental medicine and biology, 854, 307–313. https://doi.org/10.1007/978-3-319-17121-0_41
- Gliem M, Müller PL, Birtel J, Herrmann P, McGuinness MB, Holz FG & Charbel Issa P (2020). Quantitative Fundus Autofluorescence and Genetic Associations in Macular, Cone, and Cone-Rod Dystrophies. Ophthalmology. Retina, 4(7), 737–749. https://doi.org/10.1016/j.oret.2020.02.009
- Yokochi M, Li D, Horiguchi M & Kishi S (2012). Inverse pattern of photoreceptor abnormalities in retinitis pigmentosa and cone-rod dystrophy. Documenta ophthalmologica. Advances in ophthalmology, 125(3), 211–218. https://doi.org/10.1007/s10633-012-9348-8
- Birtel J, Eisenberger T, Gliem M, Müller PL, Herrmann P, Betz C, Zahnleiter D, Neuhaus C, Lenzner S, Holz FG, Mangold E, Bolz HJ & Charbel Issa P (2018). Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Scientific reports, 8(1), 4824. https://doi.org/10.1038/s41598-018-22096-0
- Garafalo AV, Sheplock R, Sumaroka A, Roman AJ, Cideciyan AV & Jacobson SG (2021). Childhood-onset genetic cone-rod photoreceptor diseases and underlying pathobiology. EBioMedicine, 63, 103200. https://doi.org/10.1016/j.ebiom.2020.103200
- Gill JS, Georgiou M, Kalitzeos A, Moore AT & Michaelides M (2019). Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. The British journal of ophthalmology, 103(5), 711–720. Advance online publication. https://doi.org/10.1136/bjophthalmol-2018-313278